骨科试验介绍3-椎体扩张球囊导管
骨科试验介绍3-椎体扩张球囊导管
乔阳
一、背景介绍
椎体扩张球囊导管主要由囊体、显影环、导管(由外管和内管组成)、连接件、单向阀/鲁尔接头(若适用)组成;部分球囊扩张导管可包含预置支撑丝,预置支撑丝由螺纹帽和导丝组成(见图1)。
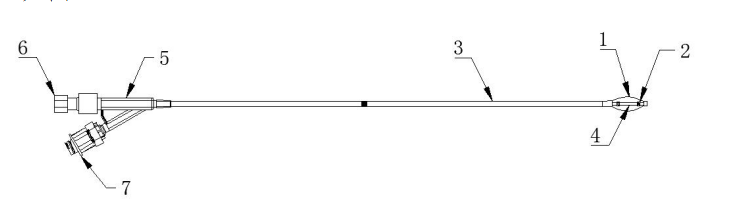
1.囊体 2.显影环 3.外管 4.内管 5.连接件
6.预置支撑丝7. 单向阀/鲁尔接头(若适用)
图1 球囊扩张导管结构示意图

a双侧撑开 b单侧撑开(定向撑开)
图2 球囊撑开后的型式
导管可为不锈钢材料或高分子材料,显影环多为铂铱合金,鲁尔接头、连接件的材料为聚碳酸酯(PC)等高分子材料。球囊的材料可为聚氨酯(PU)等。需明确各组件材料牌号。
产品工作原理
球囊扩张导管通过经皮穿刺建立椎体的工作通道,通过该通道将球囊扩张导管置于椎体内,将球囊充压装置的锁定接头与球囊扩张导管的单向阀/鲁尔接头连接,用球囊充盈装置通过显影剂为球囊进行充压,实现球囊在椎体内逐渐扩张,建立注入骨水泥的空腔。产品为一次性使用,灭菌包装。
二、临床试验
产品临床评价要求
注册申请人需按照《医疗器械临床评价技术指导原则》(国家食品药品监督管理总局通告2015年第14号)的要求选择合适的临床评价路径提交临床评价资料。
1.同品种医疗器械评价路径
详见《医疗器械临床评价技术指导原则》中通过同品种医疗器械临床试验或临床使用获得的数据进行分析评价要求。
2.临床试验评价路径
选择进行临床试验,则需严格按照《医疗器械临床试验质量管理规范》(国家食品药品监督管理总局、中华人民共和国国家卫生和计生委员会令第25号)进行临床试验,并提交完整的临床试验资料。临床试验的设计可参考《医疗器械临床试验设计指导原则》(国家食品药品监督管理总局通告2018年第6号)。
境外注册注册申请人如有境外临床试验数据,可参照《接受医疗器械境外临床试验数据技术指导原则》(国家食品药品监督管理总局通告2018年第13号)的要求提交相关临床试验资料。
对于需要在境内开展临床试验的情况,开展临床试验研究前,制定临床试验方案时建议考虑以下因素:
(1)临床试验设计类型
为了保证试验结果的真实客观性和可比性,建议采用具有良好对照的前瞻性随机对照临床试验。采用其他临床试验,如成熟设计产品可考虑单组目标值方法,申办者需提供充分的理由解释试验结果的客观性和可靠性。
(2)入选标准
PKP主要用于骨质疏松导致的椎体压缩性骨折。
①年龄范围:一般为50-80岁的患者;
②骨骼情况:疼痛性骨质疏松椎体压缩骨折,骨密度提示存在骨量丢失或骨质疏松(DXA骨密度T值≤ -2.5),主诉有明显的腰背部疼痛,无神经压迫症状;有明确责任节段(体格检查提示病变部位与影像学检查确定的节段一致);
③疾病原因:骨质疏松性压缩骨折;
④充分知情并签署知情同意书。
(4)排除标准
①椎体后壁不完整患者;
②伴有精神疾病或老年痴呆患者;
③有手术禁忌者;
④因神经受压而导致的腰背痛及相应神经症状;
⑤椎体感染或存在未控制的全身感染;
⑥高能量损伤所致创伤性椎体骨折,椎体后壁破坏广泛,较大范围不完整、后方韧带复合体损伤者;
⑦受试者合并的其他疾病限制其参加研究,不能依从随访或影响研究的科学性完整性,如患有风湿性关节炎、强直性脊柱炎患者,筛选前30天内参加了其他药物或医疗器械临床试验;
⑧拒绝签署知情同意书。
(5)受试者退出标准及退出受试者的处理
退出标准:
①受试者撤回知情同意书;
②研究者认为不再适合继续进行临床试验者;
③受试者死亡;
④受试者失访;
⑤申办者要求终止试验。
退出受试者的处理:
①尽可能记录最后一次生命体征、术后情况和局部体征检查、影像学资料和不良事件等;
②将终止试验的时间和原因详细记录在病例报告表上;
③对因不良事件而终止试验的受试者必须随访至不良事件得到解决或稳定;
④医疗器械临床试验质量管理规范规定的其他相关事宜。
(6)临床试验评价指标及判定标准
①主要评价指标:推荐使用术后6个月的有效率。将“产品有效”定义为同时满足以下要求:影像学方面,椎体矢状位Cobb角矫正率至少达到30%;术后伤椎前缘高度比值至少达到70%。疼痛缓解方面,术后VAS评分应小于3分。
a.影像学评价:
矢状位Cobb角矫正率计算公式如下:
椎体矢状位Cobb角矫正率=(术前椎体矢状位Cobb角-术后椎体矢状位Cobb角)/术前椎体矢状位Cobb角×100%;
伤椎前缘高度比值计算公式如下:
伤椎前缘高度比值=伤椎前缘高度/伤椎上下椎体前缘高度的平均值×100%。
b.疼痛评价:
采用视觉模拟评分法(visual analogue scale,VAS)评估,共计10 分,分值越低,患者疼痛程度越轻。
②次要评价指标:
患者术后腰背部疼痛及功能状态(通常采用Oswestry功能障碍指数(Oswestry disability index,ODI)进行评价)、手术时间、骨水泥注入量、止痛药物服用情况等作为次要评价指标,综合评价临床试验结果。
安全性评价指标:是否存在骨水泥渗漏或产品部件失效引起的并发症;是否存在穿刺相关神经、脊髓损伤。
(7)临床试验观察时间
作为经皮椎体后凸成形术的关键工具,需对比术前和术后的治疗效果,相关观察指标的对比需包括术前、术后即刻以及术后1个月、3个月、6个月随访的各个观察点之间的对比。临床试验的持续时间取决于所有安全性和有效性数据的获得,建议至少随访至6个月,可根据实际情况适当延长,期间设置若干个观察点。
(8)对照产品的选择
对照产品需选择目前临床正广泛使用的、对相应适应证的疗效已被证实并得到公认的、来源于同一厂家生产的产品。对照产品的材料、设计、适应证与试验产品需具有可比性。需提供对照产品的选择依据。
(9)样本量的估算
样本量一般以临床试验的主要评价指标进行估算。需在临床试验方案中说明样本量估算的相关要素及其确定依据、样本量的具体计算方法。
提供所选样本量足以评价所申报产品有效性的统计学证据,至少包括以下内容:对照组与试验组主要评价指标下的预期疗效、预期的组间差异、检验界值、显著性水平(α)、把握度(β)、预期失访率、所用到的样本量计算公式或所使用的统计学软件、建议报告引用的参考文献等。
(10)人口统计学和基线特征
①人口统计学资料:如性别、年龄、身高、体重、骨密度及骨代谢指标等;
②临床疗效相关的基线数据:考虑因素包括疾病的节段和程度、临床分类等;
③既往病史:是否有营养不良(钙、磷、蛋白质、铁)、血友病、激素缺乏(生长激素、甲状旁腺素等)、放射治疗、吸烟、嗜酒、手术史、糖尿病史、过敏病史、长期服用糖皮质激素病史等。
(11)统计分析方法
需明示具体的统计分析方法以及统计分析软件及版本。
数据分析时需考虑数据的完整性,所有签署知情同意并使用了受试产品的受试者必须纳入分析。数据的剔除或偏移数据的处理必须有科学依据和详细说明。
临床试验的数据分析需基于不同的分析集,通常包括全分析集(Full Analysis Set,FAS)、符合方案集(Per Protocol Set,PPS)和安全集(Safety Set,SS),研究方案中需明确各分析集的定义。全分析集中脱落病例,其主要研究终点的缺失值的填补方法等需足够保守并在方案中事先予以说明,必要时需进行不同分析策略的灵敏度分析,以评价缺失数据对研究结果稳定性的影响。
主要研究终点指标的分析需同时在全分析集和符合方案集上进行;安全性指标的分析需基于安全集。
临床试验数据的分析需采用国内外公认的经典统计分析方法。临床试验方案需明确统计检验的类型、检验假设、判定疗效有临床意义的界值(非劣效界值)等,界值的确定需有依据。
对验证期间发生的所有不良事件的种类、严重程度、发生频率及与验证产品的关系将列表描述。
申办者需提供基于所有临床试验数据的统计分析报告,以便临床试验牵头单位根据此报告撰写临床试验总结报告。
西格玛医学与全国脊柱外科,骨科有大量试验在开展,并已经取得大量注册证。
三、西格玛医学
南京西格玛医学技术股份有限公司(SIGMAMED),地处南京,辐射全国。西格玛医学是一家专业从事医疗器械临床研究的创新型CRO,证券代码:873450,致力于为医疗器械提供临床试验专项服务、临床研究、方案撰写、统计分析、数据管理、监查、器械SMO、受试者招募、第三方稽查和注册申报的整体解决方案。自2009年成立至今,已成功为国内外近百家客户提供专业技术服务,成功完成医学方案设计撰写、统计、稽查、临床试验、注册申报等,并建立长期稳定的合作关系。涉及主要的20余个治疗领域,骨科、眼科、肾内、护理、整形美容、IVD、血管外科、泌尿外科,麻醉科,护理部,皮肤科,心内科,心外科等,与全国20个省份近千家医院开展合作,并在国内主要省市设立一站式服务,与全国80%的临床试验机构密切合作,拥有一支中、高层稳定并按照国际标准(ICH-GCP)操作的专业团队,具有完整规范详细的标准操作规程(SOP),成功完成医疗器械Ⅱ、Ⅲ类产品临床试验1000余个,并顺利取得注册证。项目多次通过NMPA、江苏局、广东局,山东局,浙江局,四川局,福建局,北京局,天津局等和外部审核和稽查。为更好服务方便客户,我们特别开设了上海南格、深圳西格玛分公司及北京、杭州、武汉、西安等驻点办事处。